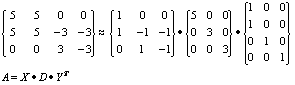
Each of the hierarchical clustering techniques within SeqExpress will establish a hierarchical graph structure which represents the differences between the different genetic profiles in the group, the resulting is a tree be visualised using the hierarchy/dendrogram viewer. SeqExpress has two different hierarchical clustering algorithms:
Semi Discrete Decomposition: builds the hierarchy by establishing how the genes contribute towards significant areas of local density (peaks/troughs).
Hierarchical Clustering: builds the hierarchy by establish which two genes are the closest together, then combining these into a single node and repeating until the tree is complete. The definition of closeness depends on a distance model, SeqExpress provides a number of distance models.
The resulting hierarchies are descended to find a number of distinct (non-overlapping) clusters, these are then added to the Gene List in the bottom right of the main SeqExpress window. Further exploration of the hierarchical relationships (and definition of different clusters) is possible by selecting the View->Dendrogram... menu option and then selecting the results.
The Semi-Discrete Decomposition (SDD) provides a quick and less computationally exhaustive method for building a hierarchy, the hierarchy can be thought of as a tri-branched decision tree (with each branch being either {1,0,-1}. SDD was originally developed for image compression. SDD attempts to identify the most significant ‘bump’ (area of local density) in a matrix, and then calculates which items are affected by it. It then removes the bumps from the matrix and attempts to find the set of next most significant bumps. This process is continued until a pre-defined number of bumps have been found, and the items which contribute towards them have been assigned. An SDD iteration involves:
In a similar manner to SVDs, the semi discrete decomposition will decompose a matrix into three separate matrices (e.g. a rotation, a scale and a rotation). e.g. Anm=XnrDrYrm’ (where A is the original matrix.).
The entries in X and Y are limited to members of the vector {1, 0,-1}: with 1 meaning the gene or experiment has a positive contribution towards the bump; 0 meaning there is no effect; and -1 meaning the effective is negative.
The above decomposition has been used to identify three factors. Examination of the X matrix shows how the different genes are involved in the ‘bumps’ that occur within the matrix. The first factor (column) is contributed towards by the first and second genes (rows) of A, the D matrix shows the first factor has a significance (height) of 5, and the Y matrix shows that the first and second experiment are significant in contributing towards the factor. The second factor (which has a significance of 3) is contributed towards by genes 2 and 3, although they contribute orthogonally. The final factor is made up of genes 2 and 3, and they both contribute in the same manner.
SDD finds a range of areas within the matrix and approximates the average of these values into one factor, thereby combining a range of complex factors into one concept. Therefore, SDD will find small clusters which represent genes which contribute towards ‘bumps’ within the expression data. Whilst the other clustering methods used in this analysis clusters find similarities in the whole of the expression pattern. The resulting X matrix can be thought of as a hierarchy (a tri-branched tree), with the distance being given by the 'singular' values in the D matrix. By descending through the tree it is possible to find clusters of genes that exhibit similar behaviour within subsets of expression data. SDD has been shown to work with high dimensional non-normal data; in particular involving sparsely populated data sets with discrete values (that is to say data that is similiar to that resulting from SAGE).
Hierarchical clustering is a commonly used and computationally expensive process. Hierarchical clustering finds the two closest nodes and then combines them into a single node, then continues to iteratively find the next two closet nodes until the tree is built. Due to the computational time involved in build such a tree, SeqExpress allows for hierarchies to be built from a sub-set of the genes.
A number of distance measures are available to generate the hierarchy (the hierarchy is based on the average distance between sets).